Nagoya, Japan - Professor Stephan Irle and Yoshio Nishimoto at the Institute of Transformative Bio-Molecules (ITbM) of Nagoya University and Dr. Dmitri Fedorov of the National Institute of Advanced Industrial Science and Technology (AIST, Tsukuba) have developed a novel ultrafast quantum chemical method enabling rapid simulations of molecules containing more than a million atoms without detrimental loss in accuracy. This method consists of a combination of the Fragment Molecular Orbital (FMO) approach and the Density-Functional Tight-Binding (DFTB) method, called FMO-DFTB and has successfully evaluated large molecules including polypeptides, a DNA segment, a small protein and a fullerite surface. The study, published online on September 22, 2014 in the journal, Journal of Chemical Theory and Computation, demonstrates a new quantum mechanical computational method achieving rapid and efficient simulation of complex molecular systems consisting of thousands to a million atoms. Simulations using FMO-DFTB could be performed on small-scale PC clusters including desktop computers, which would be useful for biologists, chemists and material scientists investigating the reactivity and structure of complex molecular systems.
The FMO-DFTB approach is a combination of FMO and DFTB methods.
(Photo Credit: ITbM, Nagoya University)
Many molecular systems encountered in biochemistry and nanoelectronics contain a large number of atoms, usually in the order of 10^3 to 10^7. Computational efforts required for quantum mechanical simulations of such systems increases steeply with the size of the system. For example, if a molecular system size increases by a factor of 100, this can result in the computational cost to be a million to a 100 trillion times more expensive depending on the method used. Therefore, simulations of large molecular systems usually require high computational expenses (computational efforts and memory requirements) and long simulation times, leading to a high demand for a rapid and accurate method to deal with such systems. The DFTB method is a semi-empirical quantum mechanical approach, which has been applied to various organic and biological systems. "Although the DFTB method is relatively fast, the computational effort increases cubically with respect to the system size. Therefore, we envisaged to combine DFTB with another approach to develop a method that can perform efficient quantum chemical calculations of complex molecular systems," says Prof. Stephan Irle, who led the research. Irle's group focused on the FMO approach, which divides complicated system into molecular fragments and has been applied to a variety of biological and inorganic systems. "By the novel combination of DFTB and FMO, we developed a unique method called FMO-DFTB, which enables ultrafast simulations with negligible error," describes Prof. Irle. Upon subjecting a water cluster system containing up to about 20,000 atoms to FMO-DFTB, the energy calculations took about 3 minutes whereas the corresponding full DFTB calculation is estimated to require about 3 weeks, which is about 10,000 times longer than FMO-DFTB. "With a nearly linear scaling of computational effort with respect to system size by the FMO-DFTB method, we have achieved a huge improvement over the DFTB approach," says Prof. Irle.
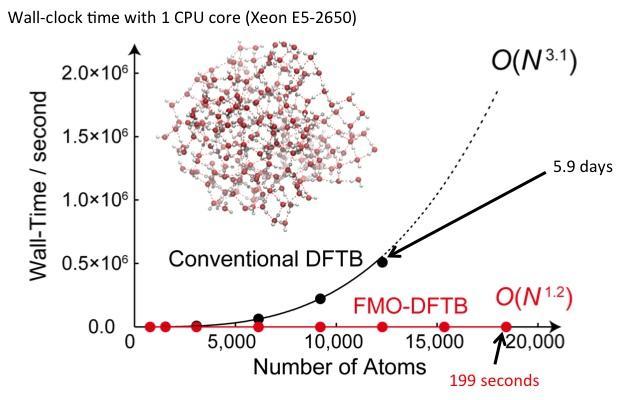
This is a graphical plot comparing the wall-clock time and scaling of conventional Density-Functional Tight-Binding (DFTB) and Fragment Molecular Orbital Density-Functional Tight-Binding (FMO-DFTB) for a water cluster (~20,000 atoms).
(Photo Credit: ITbM, Nagoya University)
The FMO-DFTB method has also demonstrated its applicability by successfully performing geometry optimization of a fullerite cluster containing more than a million atoms, which is believed to be one of the largest quantum mechanical calculations conducted up to now. "With the development of FMO-DFTB, simulations of complex molecular systems that once required supercomputers could be potentially performed on a single desktop computer, which would be useful for many people working in the fields of molecular science, along with computational theoretical scientists," says Prof. Irle. "We are currently working to further develop the accuracy of FMO-DFTB, which could be used to look into artificial peptides developed at ITbM and to explore reactions that could not have been imagined before," elaborates Prof. Irle. FMO-DFTB is considered to enable rapid simulations of large systems that were previously impossible due to high computational effort and memory requirement. Applications of FMO-DFTB are expected to extend in various areas, from chemical systems to predict reaction mechanisms pathways and biological systems to analyze ligand-protein interactions, to nanoelectronics to study the electron transport of electronic materials.

This is a photo of Prof. Stephan Irle. "With the development of FMO-DFTB, simulations of complex molecular systems that once required supercomputers could be potentially performed on a single desktop computer, which would be useful for many people working in the fields of molecular science, along with computational theoretical scientists," says Prof. Irle.
(Photo Credit: ITbM, Nagoya University)
Source: Institute of Transformative Bio-Molecules (ITbM), Nagoya University