An analysis comparing the individual differences between over 40 strains of Zika virus (30 isolated from humans, 10 from mosquitoes, and 1 from monkeys) has identified significant changes in both amino acid and nucleotide sequences during the past half-century. The data, published April 15 in Cell Host & Microbe, support a strong divergence between the Asian and African lineages as well as human and mosquito isolates of the virus, and will likely be helpful as researchers flush out how a relatively unknown pathogen led to the current outbreak.
The project--led by researchers at the University of California, Los Angeles, and the Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing--builds on other viral sequence analyses conducted over the past two months, with new large-scale and structural comparisons. Highlights of the findings include:
All contemporary human Zika virus strains share a more similar sequence to the Malaysian/1966 strain than the Nigerian/1968 strain, suggesting the strains in the recent human outbreak evolved from the Asian lineage.
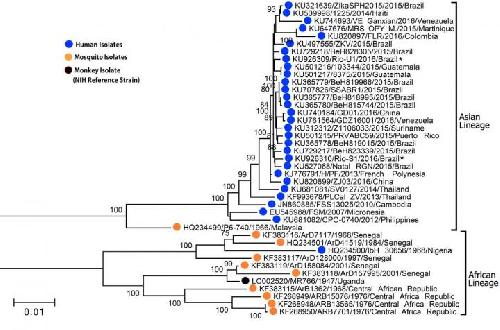 This is a phylogenetic tree constructed from nucleotide data from 41 viral complete ORF sequences of ZIKV strains by the maximum likelihood-method logarithm in MEGA7 based on the Tamura-Nei model. A bootstrap percentage for 1,000 replicates was shown on the left. Branches corresponding to partitions reproduced in less than 70% of bootstrap replicates are not shown. Strains isolated from human, mosquito, and monkey (NIH reference strain) were labeled with blue, orange, and black circles, respectively. The two subtypes were labeled on the right side of the tree. The new strains Rio-U1 and Rio-S1 were highlighted using (*). Credit: Wang and Valderramos et al./Cell Host & Microbe 2016
This is a phylogenetic tree constructed from nucleotide data from 41 viral complete ORF sequences of ZIKV strains by the maximum likelihood-method logarithm in MEGA7 based on the Tamura-Nei model. A bootstrap percentage for 1,000 replicates was shown on the left. Branches corresponding to partitions reproduced in less than 70% of bootstrap replicates are not shown. Strains isolated from human, mosquito, and monkey (NIH reference strain) were labeled with blue, orange, and black circles, respectively. The two subtypes were labeled on the right side of the tree. The new strains Rio-U1 and Rio-S1 were highlighted using (*). Credit: Wang and Valderramos et al./Cell Host & Microbe 2016
"We believe these changes may, at least partially, explain why the virus has demonstrated the capacity to spread exponentially in the human population in the Americas," says senior study author Genhong Cheng, a professor in UCLA's Department of Microbiology, Immunology & Molecular Genetics. "These changes could enable the virus to replicate more efficiently, invade new tissues that provide protective niches for viral propagation, or evade the immune system, leading to viral persistence. Of course, all of these hypotheses will need to be tested in experimental models."
Future sequencing work will likely focus on understanding the Zika strain causing the 2015-2016 epidemic, which has yet to be isolated from a mosquito. Cheng's group and others will also begin to elucidate the structure of the viral proteins, which can inform drug and vaccine design. "We hope that our work provides a strong basis that will help the larger scientific community in accelerating Zika virus research," he says.
source: Cell Press