LA JOLLA, CA—October 13, 2014—A new study by scientists from The Scripps Research Institute (TSRI), Lawrence Berkeley National Laboratory (Berkeley Lab) and other institutions suggests a cause of amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig's disease.
"Our work supports a common theme whereby loss of protein stability leads to disease," said John A. Tainer, professor of structural biology at TSRI and senior scientist at Berkeley Lab, who shared senior authorship of the new research with TSRI Professor Elizabeth Getzoff.
Getzoff, Tainer and their colleagues, who focused on the effects of mutations to a gene coding for a protein called superoxide dismutase (SOD), report their findings this week in the online Early Edition of the Proceedings of the National Academy of Sciences. The study provides evidence that those proteins linked to more severe forms of the disease are less stable structurally and more prone to form clusters or aggregates.
"The suggestion here is that strategies for stabilizing SOD proteins could be useful in treating or preventing SOD-linked ALS," said Getzoff.
Striking in the Prime of Life
ALS is notorious for its ability to strike down people in the prime of life. It first leapt into public consciousness when it afflicted baseball star Lou Gehrig, who succumbed to the disease in 1941 at the age of only 38. Recently, the ALS Association's Ice Bucket Challenge has enhanced public awareness of the disease.
ALS kills by destroying muscle-controlling neurons, ultimately including those that control breathing. At any one time, about 10,000 Americans are living with the disease, according to new data from the Centers for Disease Control and Prevention, but it is almost always lethal within several years of the onset of symptoms.
SOD1 mutations, the most studied factors in ALS, are found in about a quarter of hereditary ALS cases and seven percent of ordinary "sporadic" ALS cases. SOD-linked ALS has nearly 200 variants, each associated with a distinct SOD1 mutation. Scientists still don't agree, though, on just how the dozens of different SOD1 mutations all lead to the same disease.
One feature that SOD1-linked forms of ALS do have in common is the appearance of SOD clusters or aggregates in affected motor neurons and their support cells. Aggregates of SOD with other proteins are also found in affected cells, even in ALS cases that are not linked to SOD1 mutations.
In 2003, based on their and others' studies of mutant SOD proteins, Tainer, Getzoff and their colleagues proposed the "framework destabilization" hypothesis. In this view, ALS-linked mutant SOD1 genes all code for structurally unstable forms of the SOD protein. Inevitably some of these unstable SOD proteins lose their normal folding enough to expose sticky elements that are normally kept hidden, and they begin to aggregate with one another, faster than neuronal cleanup systems can keep up—and that accumulating SOD aggregation somehow triggers disease.
Faster Clumping, Worse Disease
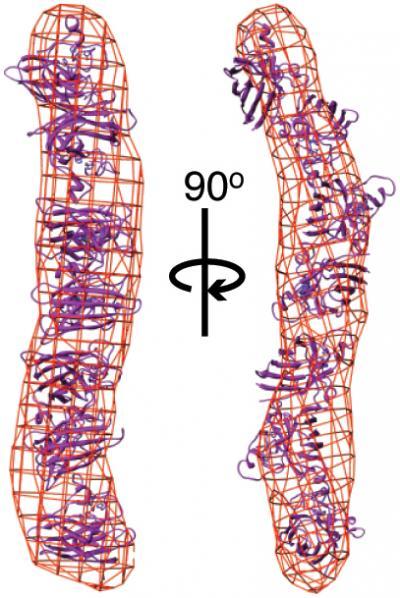
The new study provides evidence that proteins linked to more severe forms of ALS are less stable structurally and more prone to form clusters or aggregates. Mutants of the superoxide dismutase (SOD) protein formed long, rod-shaped aggregates (shown here as red lattice), compared to the compact folded structure of wild-type SOD (purple ribbons).
(Photo Credit: Image courtesy of the Getzoff and Tainer labs, The Scripps Research Institute.)
In the new study, the Tainer and Getzoff laboratories and their collaborators used advanced biophysical methods to probe how different SOD1 gene mutations in a particular genetic ALS "hotspot" affect SOD protein stability.
To start, they examined how the aggregation dynamics of the best-studied mutant form of SOD, known as SOD G93A, differed from that of non-mutant, "wild-type" SOD. To do this, they developed a method for gradually inducing SOD aggregation, which was measured with an innovative structural imaging system called SAXS (small-angle X-ray scattering) at Berkeley Lab's SIBYLS beamline.
"We could detect differences between the two proteins even before we accelerated the aggregation process," said David S. Shin, a research scientist in Tainer's laboratories at Berkeley Lab and TSRI who continues structural work on SOD at Berkeley.
The G93A SOD aggregated more quickly than wild-type SOD, but more slowly than an SOD mutant called A4V that is associated with a more rapidly progressing form of ALS.
Subsequent experiments with G93A and five other G93 mutants (in which the amino acid glycine at position 93 on the protein is replaced with a different amino acid) revealed that the mutants formed long, rod-shaped aggregates, compared to the compact folded structure of wild-type SOD. The mutant SOD proteins that more quickly formed longer aggregates were again those that corresponded to more rapidly progressing forms of ALS.
What could explain these SOD mutants' diminished stability? Further tests focused on the role of a copper ion that is normally incorporated within the SOD structure and helps stabilize the protein. Using two other techniques, electron-spin resonance (ESR) spectroscopy and inductively coupled plasma mass spectrometry (ICP-MS), the researchers found that the G93-mutant SODs seemed normal in their ability to take up copper ions, but had a reduced ability to retain copper under mildly stressing conditions—and this ability was lower for the SOD mutants associated with more severe ALS.
"There were indications that the mutant SODs are more flexible than wild-type SOD, and we think that explains their relative inability to retain the copper ions," said Ashley J. Pratt, the first author of the study, who was a student in the Getzoff laboratory and postdoctoral fellow with Tainer at Berkeley Lab.
Toward New Therapies
In short, the G93-mutant SODs appear to have looser, floppier structures that are more likely to drop their copper ions—and thus are more likely to misfold and stick together in aggregates.
Along with other researchers in the field, Getzoff and Tainer suspect that deviant interactions of mutant SOD trigger inflammation and disrupt ordinary protein trafficking and disposal systems, stressing and ultimately killing affected neurons.
"Because mutant SODs get bent out of shape more easily," said Getzoff, "they don't hold and release their protein partners properly. By defining these defective partnerships, we can provide new targets for the development of drugs to treat ALS."
The researchers also plan to confirm the relationship between structural stability and ALS severity in other SOD mutants.
"If our hypothesis is correct," said Shin, "future therapies to treat SOD-linked ALS need not be tailored to each individual mutation—they should be applicable to all of them."
The ESR experiments were performed at the laboratories of Brian Crane and Jack H. Freed at Cornell University, and the ICP-MS experiments at the laboratory of Michael W.W. Adams at the University of Georgia.
Source: Scripps Research Institute