New research from the University of Iowa answers a question that has vexed cystic fibrosis (CF) researchers for almost 25 years: why don't mice with CF gene mutations develop the life-threatening lung disease that affects most people with CF?
Published Jan. 29 in the journal Science, the research led by Michael Welsh, M.D., provides an answer to this long-standing scientific puzzle, and in doing so, identifies a proton pump that may be a target for new CF therapies.
"Since the first CF mouse was reported in 1992, I have been asked hundreds of times, 'Why don't CF mice have respiratory host defense defects and develop lung infections?'" says Welsh, UI professor of internal medicine, molecular physiology and biophysics, a Howard Hughes Medical Institute Investigator, and Director of the Pappajohn Biomedical Institute.
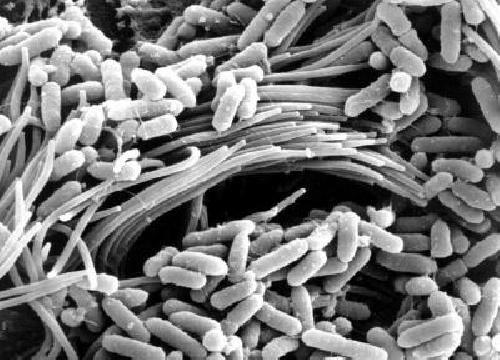 This is a scanning electron microscopy image shows bacteria (Pseudomonas aeruginosa) on the surface of a CF-affect airway. Abnormally high acidity in CF airways impairs the ability of the airways to fight off bacterial infection. A new study identifies a proton pump called ATP12A as a cause of increased acidity in CF airways of people and pigs. The protein is missing in mice. The finding provides an explanation of why CF mice do not get lung disease and suggests that ATP12A may be a target for new CF therapies. Credit: Tom Moninger, University of Iowa Health Care
This is a scanning electron microscopy image shows bacteria (Pseudomonas aeruginosa) on the surface of a CF-affect airway. Abnormally high acidity in CF airways impairs the ability of the airways to fight off bacterial infection. A new study identifies a proton pump called ATP12A as a cause of increased acidity in CF airways of people and pigs. The protein is missing in mice. The finding provides an explanation of why CF mice do not get lung disease and suggests that ATP12A may be a target for new CF therapies. Credit: Tom Moninger, University of Iowa Health Care
In answering this question, Viral Shah, first author of the study and a student in the Medical Scientist Training Program at the UI Carver College of Medicine, homed in on the thin layer of liquid that covers the airways, i.e., the tracheal and bronchial passages. Shah and his colleagues specifically studied the liquid's acidity. The importance of acidity was revealed in earlier UI studies using CF pigs. That work showed that CF pigs have an abnormally acidic airway liquid, and that increased acidity impairs the ability of the airways to fight off infection.
Shah explained that normally, two opposing processes control airway acidity. The cystic fibrosis transmembrane conductance regulator (CFTR) channel secretes bicarbonate, a base. That process is countered by secretion of protons, an acid. The balance tightly controls the acidity of liquid in the airways.
In people, pigs, and mice with CF, the CFTR channel is lost, stopping the flow of bicarbonate into airways. When that happens in people and pigs, the airway liquid becomes more acidic, degrading the ability to fight infection. But in mice, the airway liquid does not become more acidic and they are not prone to infection. That led the scientists to ask what secretes acid into airways of people and pigs, but is missing in mice. They discovered that a proton pump called ATP12A is responsible.
Shah and his colleagues made the discovery by comparing airway tissue from humans, pigs, and mice. The scientists showed that blocking ATP12A in airway tissue from pigs and humans with CF reduces the acidity of airway liquid and restores the airway's defenses against infection. Conversely, putting the ATP12A proton pump into airways of CF mice increases acidity of the liquid and predisposes the CF mice to bacterial infections.
"This discovery helps us understand the cause of lung disease in people with CF. It may also identify ATP12A as a new therapeutic target," Shah says. "We wonder if blocking ATP12A in people with CF could halt the progression of lung disease."
Shah added that targeting ATP12A could potentially be helpful for all forms of CF regardless of a patient's CFTR mutation because ATP12A is independent of CFTR.
The CF pig model was developed in 2008 by Welsh and his research team at the UI with colleagues from the University of Missouri. The CF pig closely mimics human CF disease, including the lung problems that are missing in CF mice, and has proven very useful in advancing understanding of CF lung problems.
source: University of Iowa Health Care