ALBUQUERQUE, N.M. — Sandia National Laboratories researcher Steve Plimpton, who led development of a widely used computer code that models how materials behave, has been invited to present a keynote lecture at the Feb. 27-March 3 Minerals, Materials & Materials Society (TMS) meeting in San Diego.
Plimpton developed the LAMMPS molecular-dynamics software code. The acronym LAMMPS (Large-scale Atomic/Molecular Massively Parallel Simulator) is also a pun on the word "lamp," a device that brings light to dark places.
"This symposium [on Massively Parallel Simulations of Materials Response] will particularly recognize your contribution as the primary developer of one large-scale parallel code, the LAMMPS molecular dynamics simulator, which has been of great help to the technical community, wrote symposium co-organizer and Virginia Tech engineering professor Diana Farkas in her invitation to Plimpton. "As users of LAMMPS, we feel that this contribution really deserves a special recognition."
LAMMPS is an open-source code (lammps.sandia.gov) initially funded fifteen years ago through an industrial Cooperative Research and Development Agreement (CRADA) between Sandia and multiple collaborators. Subsequent additions were supported by Sandia's Laboratory Directed Research and Development program, various Department of Energy (DOE) offices, and currently a recent CRADA focused on modeling nanoparticles in solution. Over the years, more than 100 significant contributions to the code have been made by Plimpton and other Sandia and external researchers.
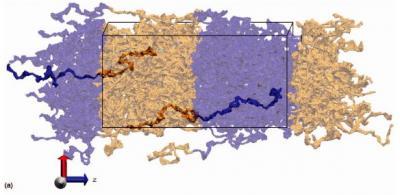
French researchers Michel Perez, Olivier Lame, Fabien Leonforte, and Jean-Louis Barrat used a versatile method, largely inspired by chemical radical polymerization, to generate configurations of coarse-grained models for polymer melts.
(Photo Credit: Images courtesy of Sandia National Laboratories)
At the atomic scale, LAMMPS can model soft (biomolecules, polymers) or solid-state (metals, semiconductors) materials. Using a more coarse-grained approach, the program can also model mesoscopic or even macroscopic systems as a collection of interacting particles. Able to run on single processors or multiple parallel processors, the code is designed to be easy to modify or extend with new functionality.
This ease of use has led to some 65,000 downloads since its open-source release in 2004, and more than 1,000 journal articles that have cited the code.
Due to its parallel scalability, LAMMPS often helps benchmark performances of the newest DOE and Department of Defense supercomputers, including the DOE Leadership Computing Facility at Oak Ridge National Laboratory. To benchmark the next-generation LCF computer, which is expected to have a hybrid CPU/GPU [central processing unit/graphics processing unit] design, ORNL is supporting a multi-institutional team, led last year by Sandia researcher Paul Crozier, to extend LAMMPS to run on general-purpose GPUs.
Crozier is a LAMMPS co-developer, as are Sandia researchers Aidan Thompson and Mark Stevens.
A year ago, said Sandia manager John Aidun, the first LAMMPS users' meeting was held at Sandia.
"This bare-bones, invitation-only meeting (due to space limitations) was well attended by enthusiastic users and contributors to LAMMPS," he said. "It demonstrated that the code has become an institution, with at least one DoD lab having discontinued its own code development efforts and now depending on LAMMPS to meet its mission responsibilities."

Researchers from Northwestern University and Sandia, working with the LAMMPS code, developed a simulation strategy for solidifying metals and metal alloys where the temperature of the system is carefully thermostatted so the velocity of the interface can be accurately measured. The image here depicts a liquid/solid interface in a nickel /aluminum alloy.
(Photo Credit: Images courtesy of Sandia National Laboratories)
Source: DOE/Sandia National Laboratories