Nature, through the trial and error of evolution, has discovered a vast diversity of life from what can only presumed to have been a primordial pool of building blocks.
Inspired by this success, a new Biodesign Institute research team, led by John Chaput, is now trying to mimic the process of Darwinian evolution in the laboratory by evolving new proteins from scratch. Using new tricks of molecular biology, Chaput and co-workers have evolved several new proteins in a fraction of the 3 billion years it took nature.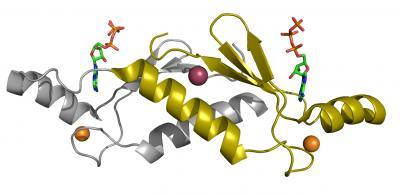 The three-dimensional structure (ribbon diagram) of protein DX as a crystallogrpahic dimer. The zinc metal ion is shown in orange and the chloride anion at the dimer interface in red, and the ATP ligand is colored by atom type. DX has been evolved in the lab to improve protein stability and can bind ATP tighter than similar proteins found in nature. Credit: John Chaput, the Biodesign Institute at Arizona State University
The three-dimensional structure (ribbon diagram) of protein DX as a crystallogrpahic dimer. The zinc metal ion is shown in orange and the chloride anion at the dimer interface in red, and the ATP ligand is colored by atom type. DX has been evolved in the lab to improve protein stability and can bind ATP tighter than similar proteins found in nature. Credit: John Chaput, the Biodesign Institute at Arizona State University
Their most recent results, published in the May 23rd edition of the journal PLoS ONE, have led to some surprisingly new lessons on how to optimize proteins which have never existed in nature before, in a process they call ‘synthetic evolution.’
"The goal of our research is to understand certain fundamental questions regarding the origin and evolution of proteins," said Chaput, a researcher in the institute’s Center for BioOptical Nanotechnology and assistant professor in Arizona State University’s department of chemistry and biochemistry. "Would proteins that we evolve in the lab look like proteins we see today in nature or do they look totally different from the set of proteins nature ultimately chose" By gaining a better understanding of these questions, we hope to one day create new tailor-made catalysts that can be used as therapeutics in molecular medicine or biocatalysts in biotechnology."
The building blocks of proteins are 20 different amino acids that are strung together and folded to make the unique globular shape, stability and function of every protein. The mixing and matching of the amino acid chain like numbers in the lottery are what favor the odds in nature of finding just the right combinations to help generate biological diversity. Yet no one can predict how the string of amino acids sequence folds to make the 3-D functional structure of a protein.
To select the raw ingredients to create the proteins, Chaput’s group (which includes Harvard collaborator Jack Szostak, and ASU colleagues Jim Allen, Meitian Wang, Matthew Rosenow and Matthew Smith) began their quest by further evolving a protein that had been previously selected from a pool of random sequences.
Jack Szostak and Anthony Keefe first made the parental protein in 2001. To achieve their feat, they stacked the odds of finding just one or two new proteins and generated a library of random amino acid sequences so vast — 400 trillion — that it dwarfs the number of items in the entire Library of Congress (134 million).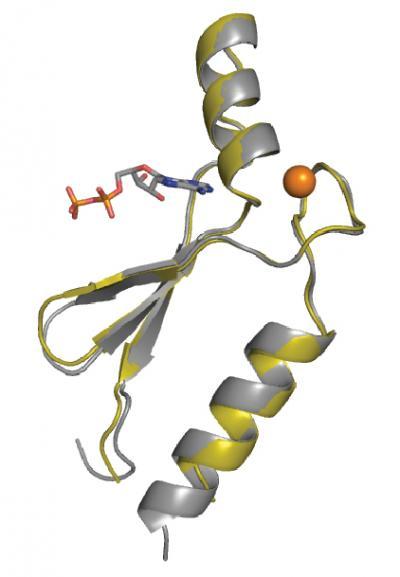 The 3-D structure (ribbon diagram) of protein DX (gray) superimposed with the parent sequence, protein 18-19 (gold). The zinc metal ion is shown in orange and the ATP ligand is colored by atom type. Credit: John Chaput, Biodesign Institute at Arizona State University
The 3-D structure (ribbon diagram) of protein DX (gray) superimposed with the parent sequence, protein 18-19 (gold). The zinc metal ion is shown in orange and the ATP ligand is colored by atom type. Credit: John Chaput, Biodesign Institute at Arizona State University
They started with a small protein stretch 80 amino acids long. This basic protein segment acts as a protein scaffold that can be selected for the ability to strongly clutch its target molecule, ATP.
There was only one problem, the parental protein could bind ATP, but it wasn’t very stable without it.
"It turns out that protein stability is a major problem in biology," said Chaput. "As many as half of the 30,000 genes discovered from the human genome project contain proteins that we really don’t know what their structure is or whether or not they would be stable. So for our goal, we wanted to learn more about the evolution of protein folding and stability."
Chaput’s group decided to speed up protein evolution once again by randomly mutating the parental sequence with a selection specically designed to improve protein stability. The team upped the ante and added increasing amounts of a salt, guanidine hydrochloride, making it harder for the protein fragment to bind its target (only the top 10 percent of strongest ATP binders remained). After subjecting the protein fragments to several rounds of this selective environmental pressure, only the ‘survival of the fittest’ ATP binding protein fragments remained.
The remaining fragments were identified and amino acid sequences compared with one another. Surprisingly, Chaput had bested nature’s designs, as the test tube derived protein was not only stable, but could bind ATP twice as tight as anything nature had come up with before.
To understand how this information is encoded in a protein sequence, Chaput and colleagues solved the 3-D crystal structures for their evolutionary optimized protein, termed DX, and the parent sequence.
In a surprising result, just two amino acids changes in the protein sequence were found to enhance the binding, solubility and heat stability. "We were shocked, because when we compared the crystal structures of the parent sequence to the DX sequence, we didn’t see any significant changes," said Chaput. "Yet no one could have predicted that these two amino acids changes would improve the function of the DX protein compared to the parent.
The results have helped provide a new understanding of how subtle amino acid changes contribute to the protein folding and stability. Chaput’s team has developed the technology potential to take any of nature’s proteins and further improve its stability and function. "We have the distinct advantage over nature of being able to freeze the evolution of our lab-evolved proteins at different time points to begin to tease apart this random process and relate it to the final protein function," said Chaput.
Next, Chaput plans on further expanding his efforts to evolve proteins with new therapeutic features or catalytic functions.
Source: Arizona State University